
Pick’s Disease (PiD)
PiD is a very rare neurodegenerative disorder clinically characterized by dementia, with frontotemporal degeneration and tau intracellular inclusions known as Pick bodies.
Dr. Arnold Pick introduced the disease in 1892
PiD was first described in 1892 by German physician Dr. Arnold Pick, who observed and reported on the clinical symptoms of a patient presenting with memory impairment and language difficulties. However, at the time, Pick did not realize he was describing a new neurodegenerative disease. In the following years, reported more cases with similar clinical symptoms, atrophy of the brain regions involved in language and behavior, and pathological features distinct from Alzheimer’s disease. While Pick’s reports were detailing the clinical presentation of PiD patients with some minimal neuropathological findings, it wasn’t until 1911 that Pick bodies were first visualized by Dr. Alois Alzheimer who characterized the two hallmarks of PiD pathology, namely argyrophilic inclusions (Pick bodies) and swollen cells (Pick cells). In 1923, Pick’s students introduced the term “Pick atrophy” to describe cases with atrophy of the frontal and temporal lobes of the brain, which quickly became a term used to describe all cases with frontotemporal lobar degeneration. It wasn’t until a few years later, in 1926, that the term Pick’s disease was coined to describe this new non-AD dementia.
Disease Background
The term “Pick’s disease” has been used to describe many different diseases over the years
When Pick’s disease became a separate disease entity, in 1926, the term PiD was used to describe all dementia cases that had frontotemporal degeneration upon brain autopsy and pathology distinct of that of Alzheimer’s disease. In fact, the term PiD became almost synonymous to the broader term frontotemporal degeneration, contributing to inconsistencies in the literature. Attesting to that, in 1974, Dr. Constantinidis defined three neuropathological subtypes of PiD, Type A, Type B, and Type C, all describing separate pathologies.
Around the same time, the scientific community started focusing on AD neuropathology and PiD research was kept alive by two groups: one in Lunch, Sweden, led by Drs. Gustafson, Ingvar, and Brun, and the other in Manchester, England, led by Drs. Neary, Snowden, and Mann. The two groups came together in 1994 to publish a comprehensive guide of neuropathological and clinical criteria for non-AD cases presenting with atrophy of the frontal and temporal lobes. They were the first to distinguish three subtypes of frontotemporal dementia (FTD); namely, frontal lobe degeneration type, Pick type, and Motor neuron disease type, clearly classifying PiD as a pathology term and a variant of FTD.
Many revisions in terminology have been made since then which have all culminated in the creation of a Frontotemporal Lobar Degeneration spectrum of disease (FTLD) encompassing all pathological diagnoses of the clinical FTD syndromes.
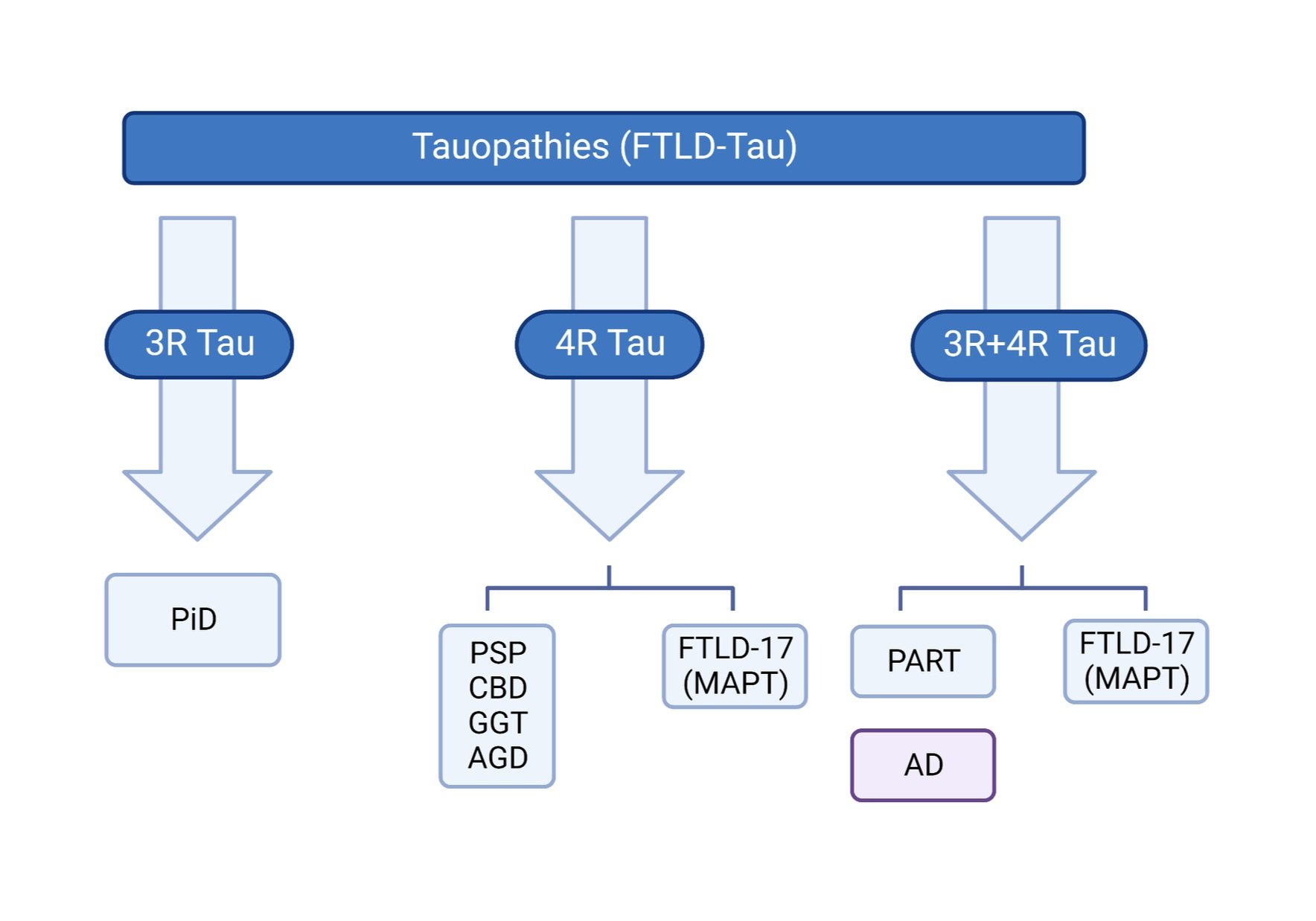
PiD is part of the FTLD spectrum and the only known primary 3R tauopathy
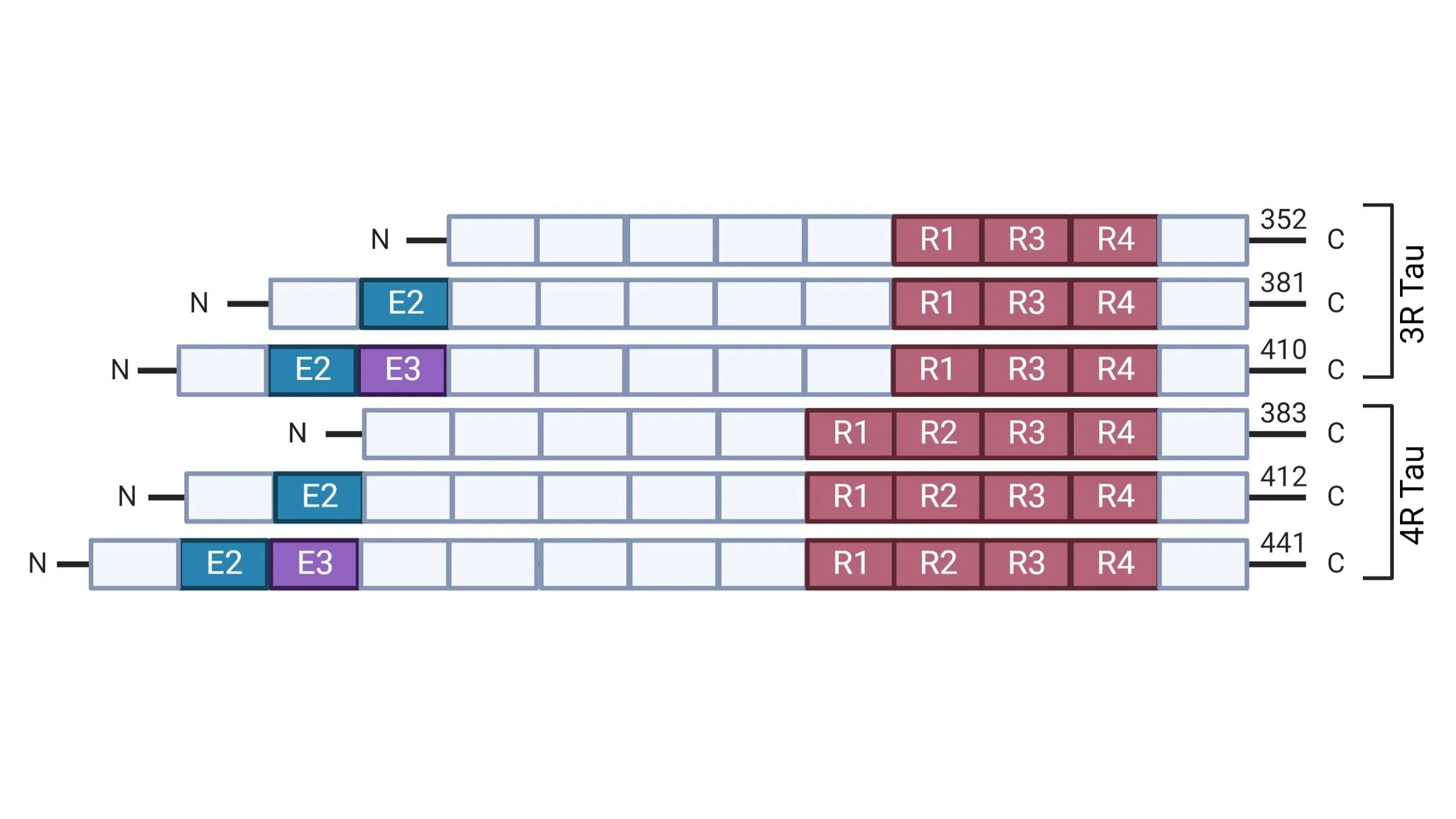
PiD is a type of primary tauopathy along with Progressive Supranuclear Palsy (PSP), Corticobasal Degeneration (CBD) and Primary Age Related Tauopathy (PART). Primary tauopathies can be further classified as 3-Repeat (3R; PiD), 4-Repeat (4R; PSP, CBD), and 3R+4R (PART) based on the predominance of the tau isoform present in the aggregates characterizing each disease. More specifically, 3R and 4R tau isoforms differ in the presence (4R) or absence (3R) of MAPT exon 10 which encodes the second repeat in the microtubule binding domain of the protein. In the adult human brain, the 3R and 4R groups of isoforms exist in equilibrium; however, in various neurodegenerative diseases we observe preferential increase and accumulation of specific isoforms. Interestingly, PiD is the only disease known to be characterized by preferential aggregation of 3R tau isoforms.
Pick significantly contributed to the research of dementias with his hypothesis that dementia is a result of the summation of atrophy at different brain regions; an innovative idea at a point in time when dementia was thought to be a diffuse process caused by senility or vascular disease.

Created with BioRender.com
Neuropathologically, PiD is characterized by severe, atrophy of the frontal and temporal lobes. The atrophy extends to subcortical areas including the amygdala, entorhinal cortex, and cingulate gyrus. However, the hippocampus remains relatively preserved and substantia nigra well pigmented, distinguishing PiD from other neurodegenerative disorders. PiD brains are also characterized by severe gliosis across the affected cortical areas.
Neuropathology
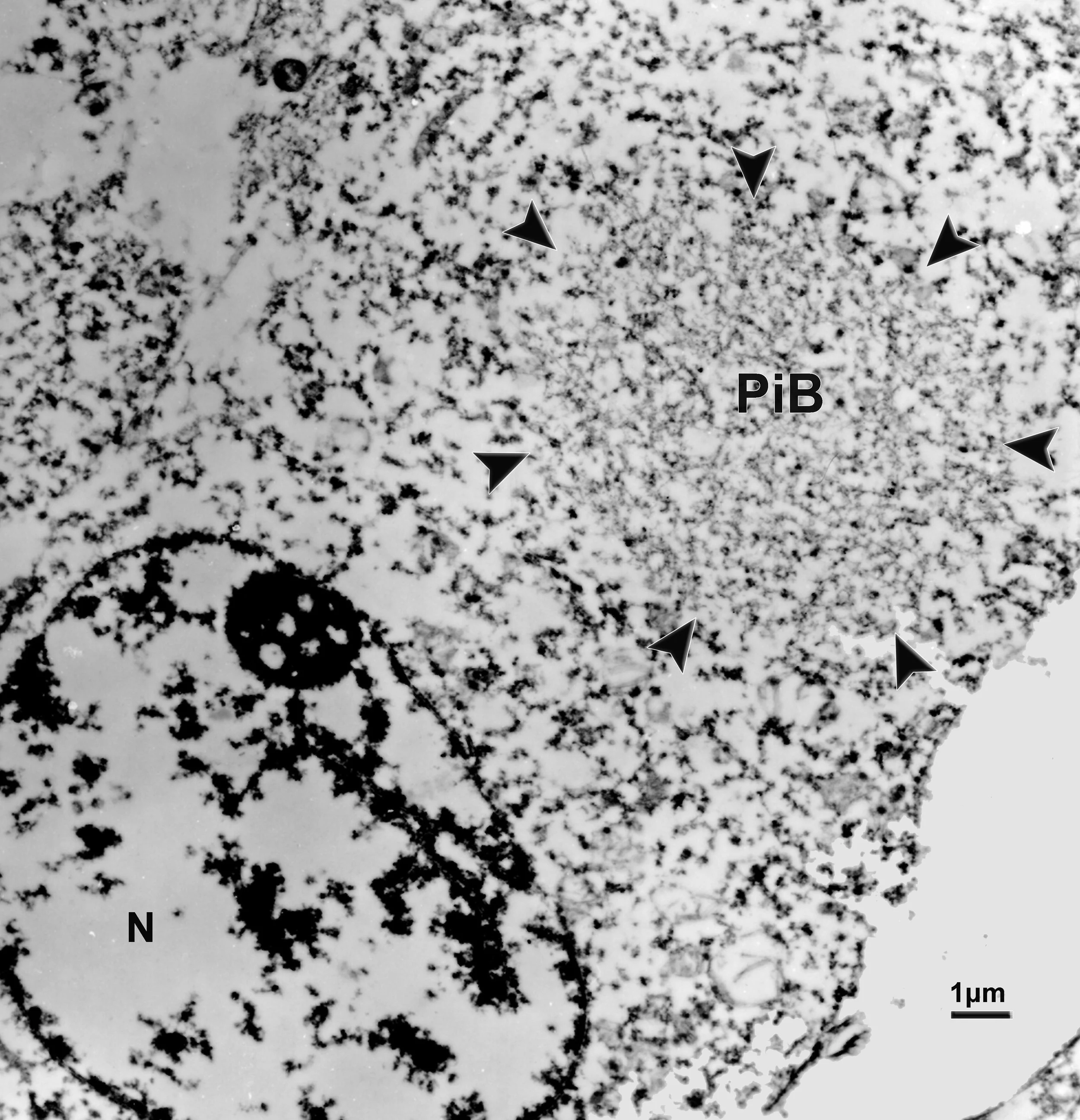
The Pick Body: the hallmark of PiD neuropathology
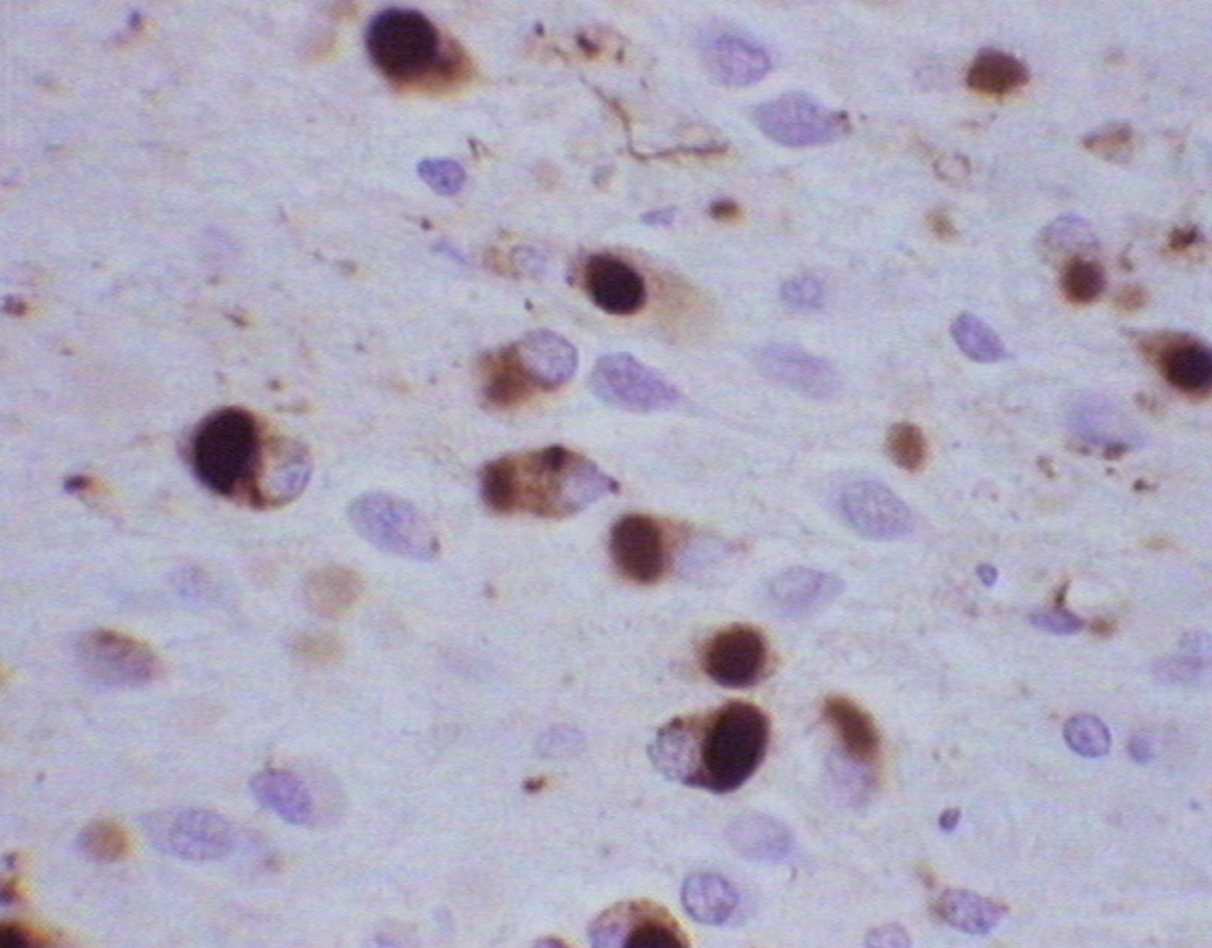
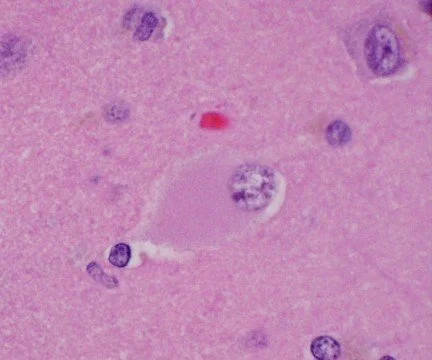
The hallmark of PiD disease neuropathology is the presence of swollen neurons termed Pick cells and intracellular tau aggregates known as Pick bodies. Pick cells are ballooned neurons found throughout the periphery of affected cortical areas. Pick bodies are spherical argyrophilic neuronal cytoplasmic inclusions that are most prominent in the hippocampus but can also be found in certain layers of the cortex. Pick bodies are negative for α-synuclein which readily differentiates them from Lewy bodies and morphologically distinct from other tau aggregates seen in disease conditions, like neurofibrillary tangles in Alzheimer’s disease. However, little is known about the exact composition of Pick bodies.
Clinical Symptoms
PiD has an average age of onset of 55 years of age and it affects women and men equally.
PiD patients will most frequently present with executive dysfunction, lack of empathy, disinhibition as well as speech disturbances such as apraxia of speech.
The prominent behavioral changes observed in PiD patients early on in disease progression can sometimes resemble symptoms of various neuropsychiatric disorders, including schizophrenia or bipolar disorder.
Since a PiD diagnosis can only be given postmortem, upon brain autopsy, most PiD patients are clinically diagnosed with either behavioral variant FTD (bvFTD) or primary progressive aphasia (PPA). However, clinical diagnosis of corticobasal syndrome (CBS) or Alzheimer’s disease (AD) are also common.
PiD can only be diagnosed post mortem, upon brain autopsy. For a definitive PiD diagnosis, a given case must meet strict neuropathological and biochemical criteria.