
Ongoing Genetics Studies
Pick’s disease is a primary 3R tauopathy characterized by aggregation of misfolded 3R tau protein in cytoplasmic inclusions called Pick bodies. Thus, we are trying to characterize the relationship between tau and disease risk, pathogenesis, and progression at the genetic level.
The MAPT region
The tau protein is encoded by the MAPT gene on chromosome 17. The role of MAPT variation in neurodegenerative diseases has been extensively studied in the context of Alzheimer’s disease, Frontotemporal Dementia , and other 4R tauopathies (Progressive Supranuclear Palsy, Corticobasal degeneration), but remains poorly understood in Pick’s disease. In fact, MAPT variation has been shown to be sufficient for dementia and neurodegeneration with tau pathology. This has been demonstrated by numerous studies of affected families with neurodegenerative disease consistent with Frontotemporal Dementia and underlining 4R tau pathology (FTLD-17).
In addition, MAPT falls within the largest block of linkage disequilibrium in the human genome spanning approximately 1.8Mb. This region is divided into two haplotypes: H1 and H2, which are defined by numerous single nucleotide polymorphisms (SNPs) and a 900kb inversion that contains the MAPT gene as well as neighboring genes IMP5, CRHR1, and NSF. The H1 MAPT haplotype is the major risk factor for 4R tauopathies (Progressive Supranuclear Palsy, Corticobasal degeneration), and has also been shown to be associated with increased risk of other neurodegenerative diseases like Parkinson’s disease. While the H2 MAPT haplotype is thought to be protective. Few earlier studies have been underpowered to detect an association of either of the haplotypes with Pick’s disease risk.
We are investigating the relationship between the MAPT haplotypes and Pick’s disease risk, in the largest Pick’s disease cohort described to date.
GWAS efforts
In addition to the investigation of MAPT specific variation (haplotypes) as a contributor to Pick’s disease risk, the PIC is also carrying out genome wide association studies (GWAS) to define other potential common variation. Through these efforts we will be able to nominate common genetic variation, outside the MAPT region, that are contributing to disease risk and clinical presentation.
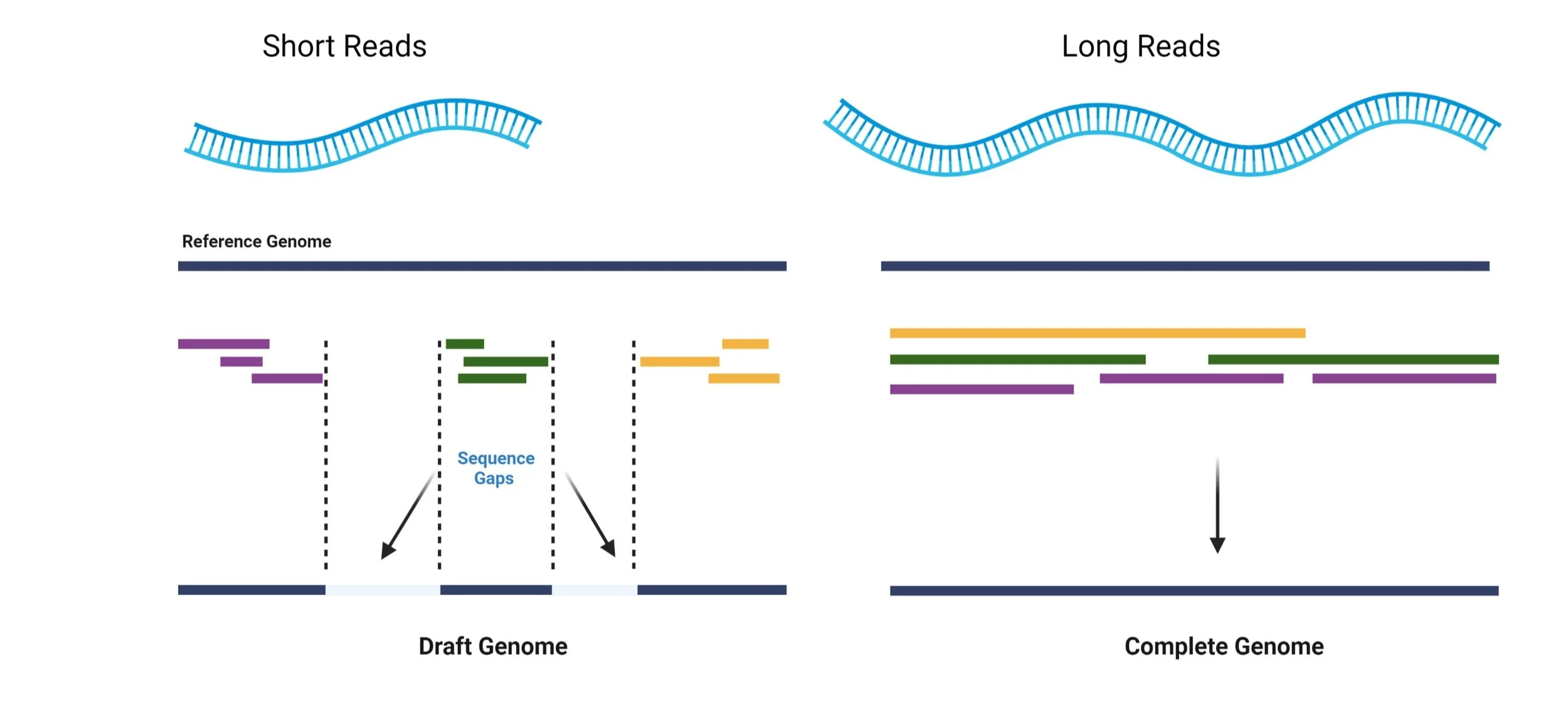
The Future: Long-Read Whole Genome Sequencing
In the future, we will expand our studies to investigate rare and complex variation in the genome that could be conferring Pick’s disease risk or influencing the clinical symptoms of the disease. More specifically, by employing long-read whole genome sequencing approaches we will be able to characterize variation in regions of the genome that do not achieve good coverage with traditional next-generation sequencing technologies and as a result remain understudied. These regions include repeat or GC-rich domains, pseudogenes, as well as large and complex structural variation like inversions, deletions and duplications that could have critical consequences on proper gene function.
Created with BioRender.com